FragmentLab is a simple but effective application for visualizing and analyzing peptide fragmentation spectra. It was originally developed to test the code for our crosslinked peptide search engine XlinkX/PD, but has since come into its own and has been useful for many projects and its results have featured in many publications,
Generic upload
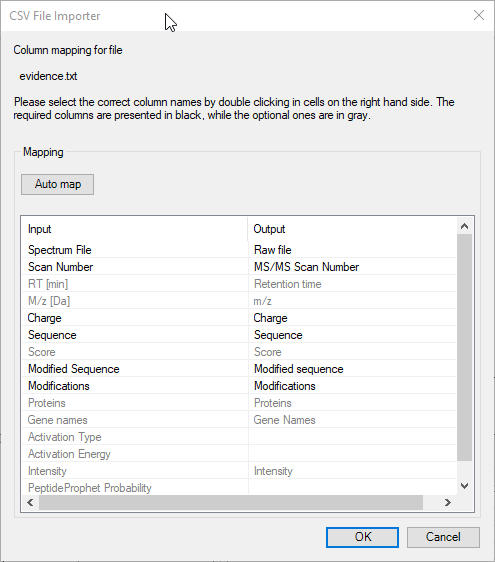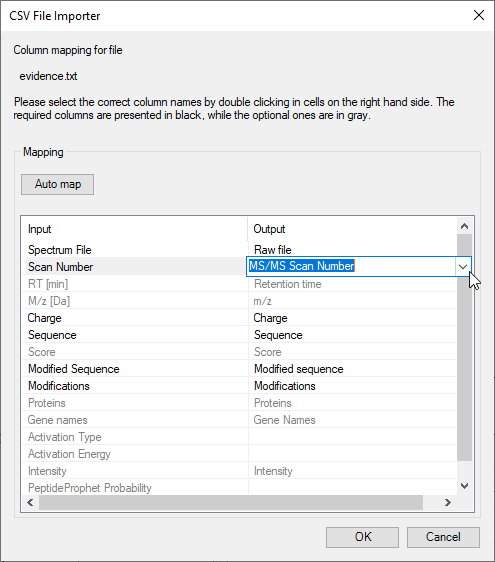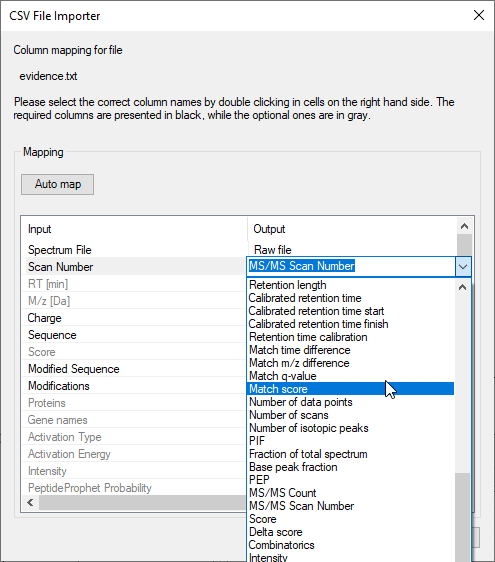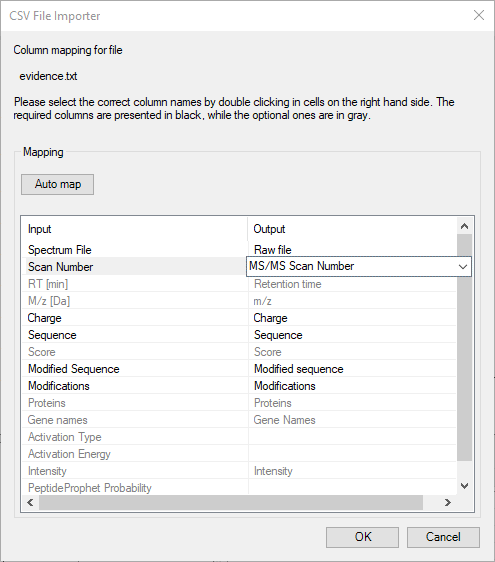
FragmentLab aims to offer generic upload of results. Although this will be out of reach as each search engine has different notations for many things, this attitude comes in handy as all search engines have their own peculiarities. For example MaxQuant cheekily changes column names in the output formats between versions.
After File → Open and selecting the relevant result file, FragmentLab will read the column names and make an intelligent guess of their purpose. The results are presented in an editor where you can still make changes if required.
Modification editor
Even though FragmentLab carries an extensive list of modifications, it can be that yours is not there or you would like to include additional fragmentation behavior.
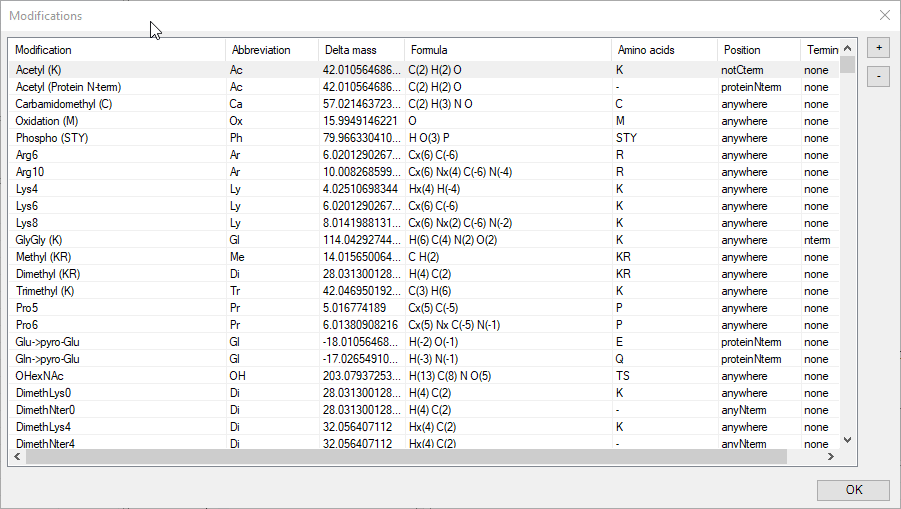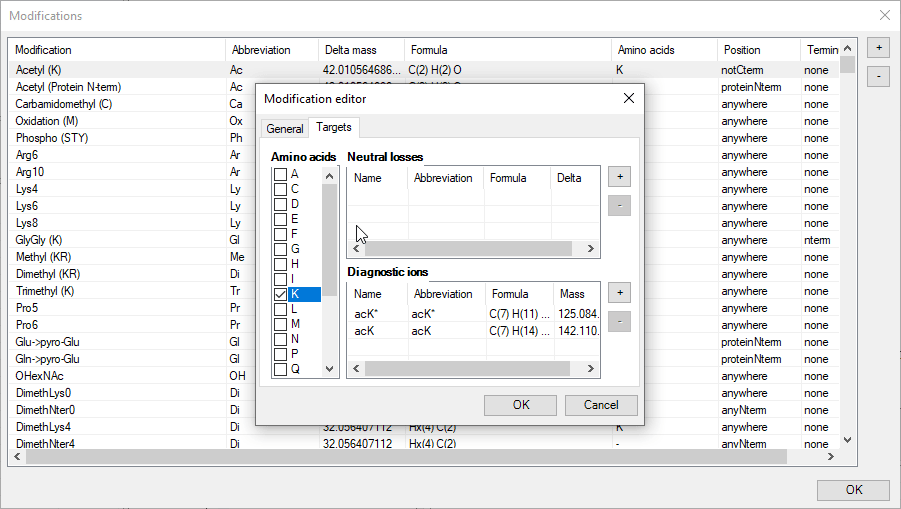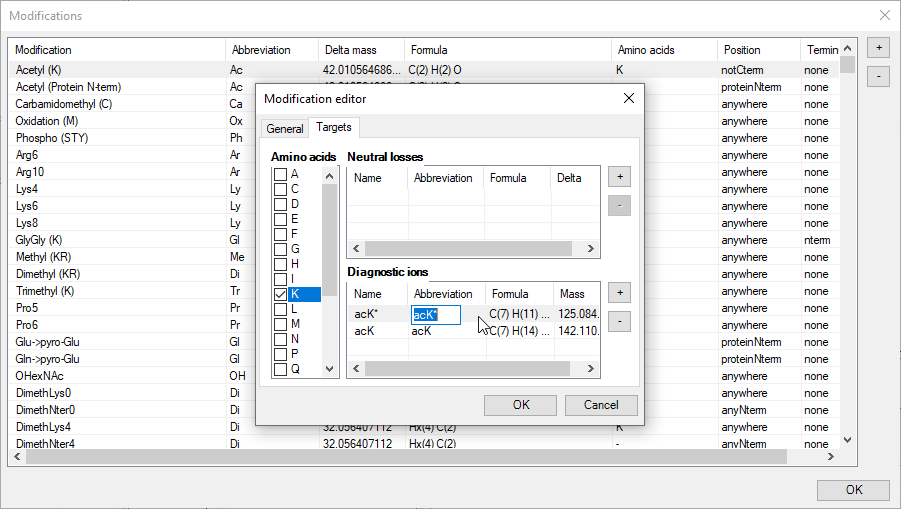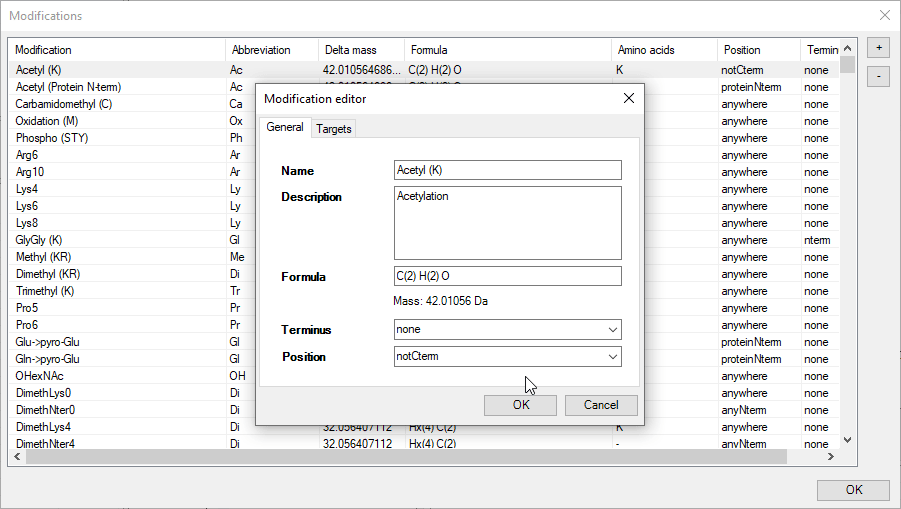
An intuitive editor is provided for precisely this purpose. New modifications can be added with the + button and all modifications can be edited. This includes:
1. Naming;
2. Chemical formula;
3. Whether it is located on the terminus;
4. Position information;
5. Neutral losses;
6. Diagnostic ions.
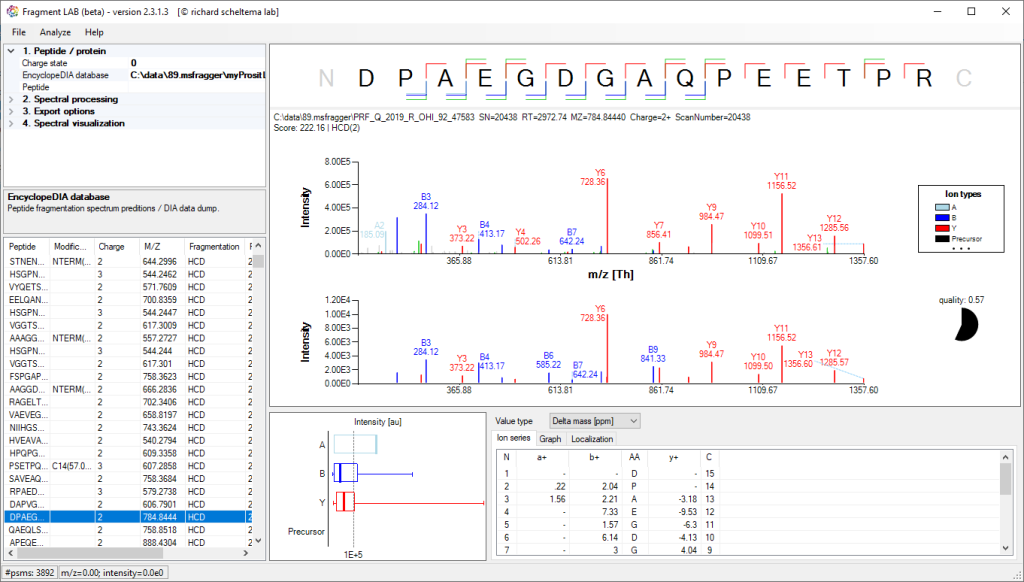
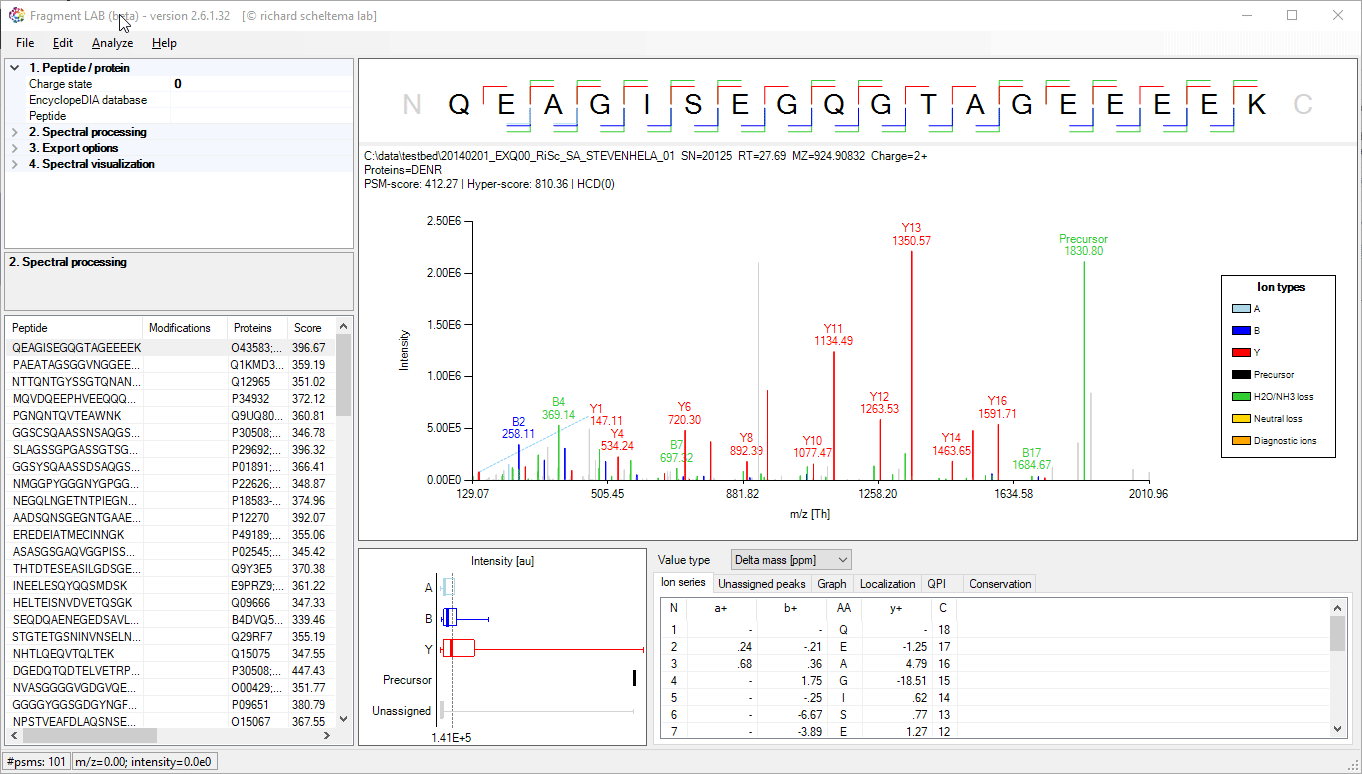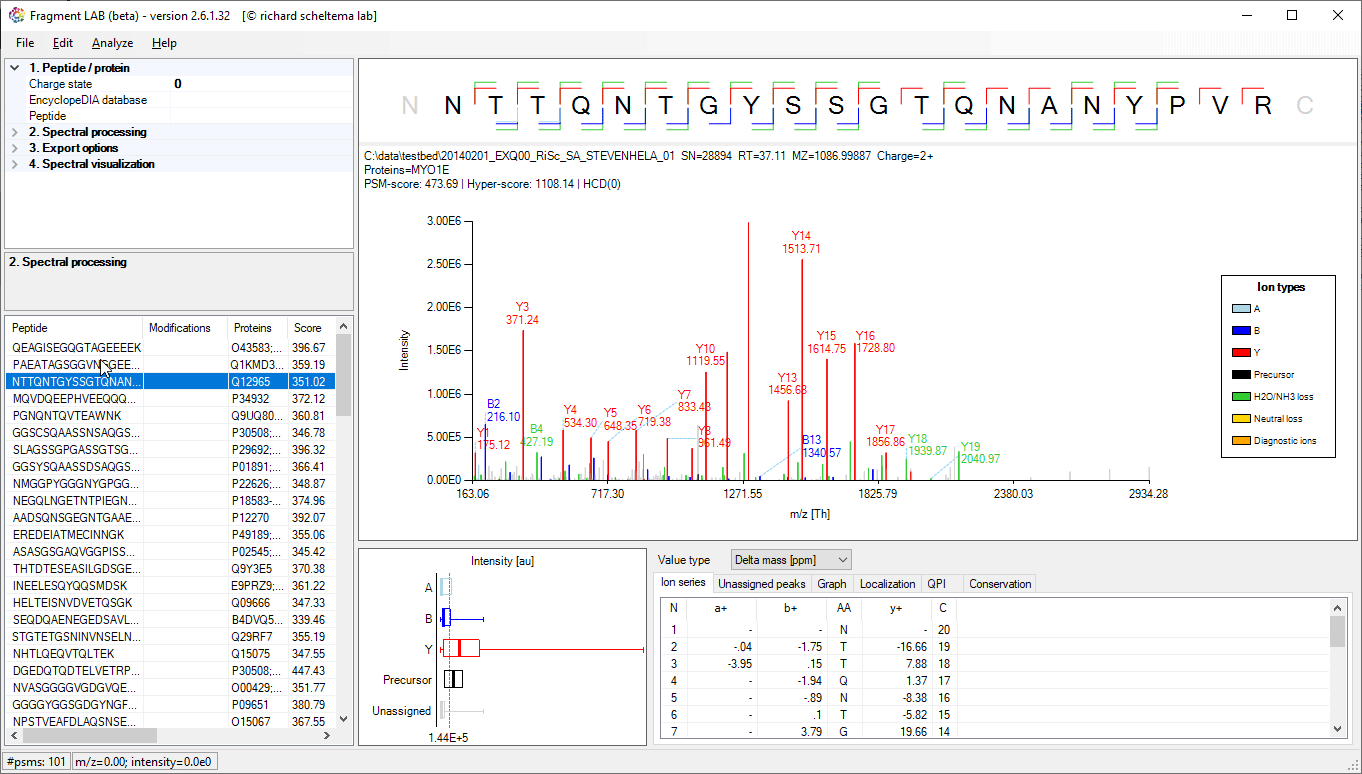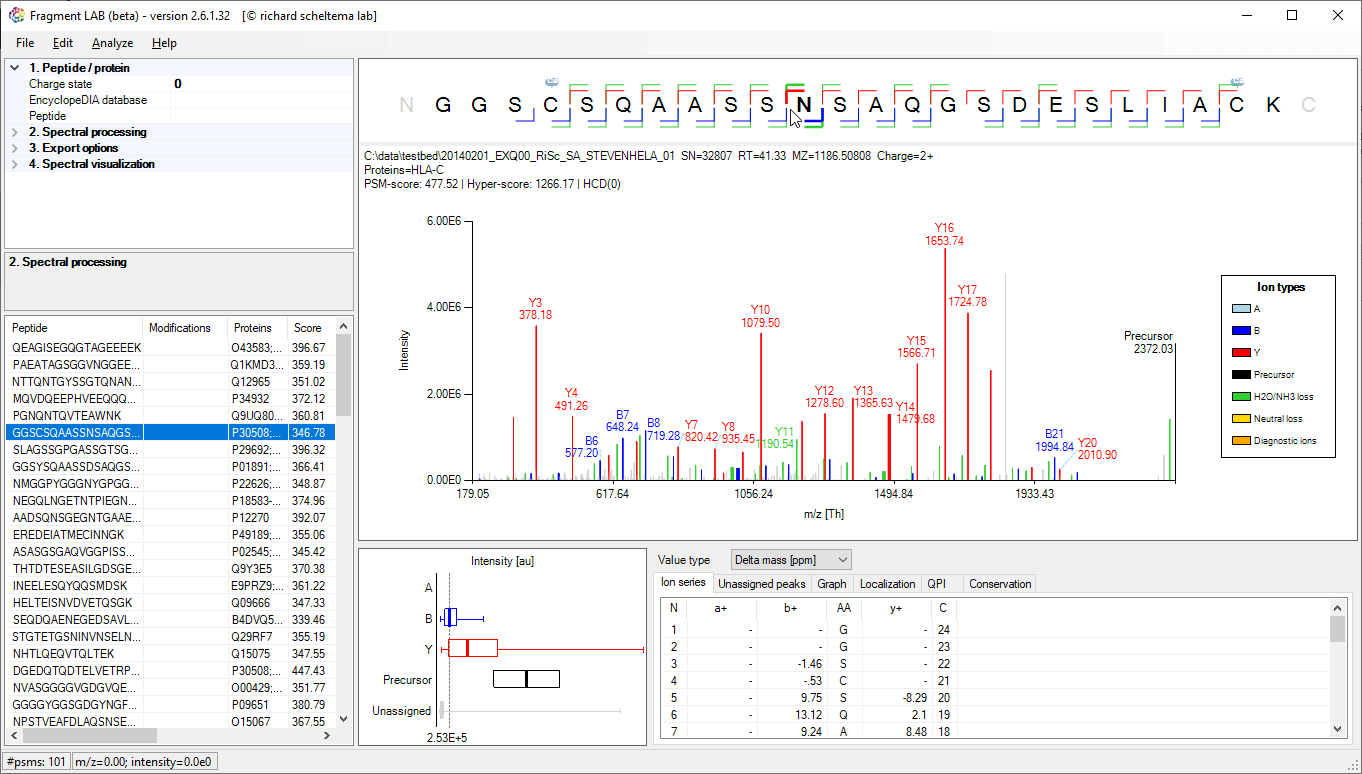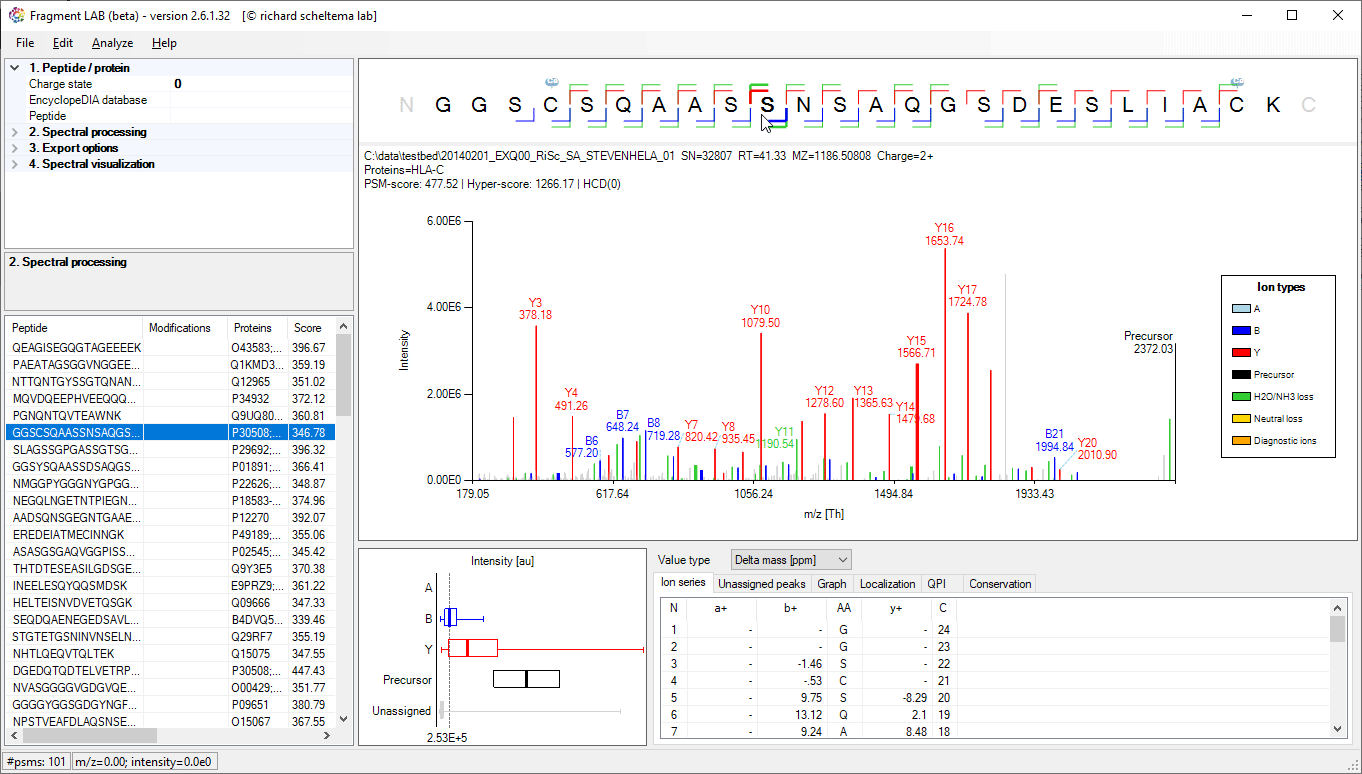
Spectrum annotations
After loading the result file, it is easy to inspect the fragmentation spectra by selecting entries in the PSM table on the bottom left. After visualization, the spectrum can be further inspected in detail to find the peaks associated with an amino acid position and locating the m/z precision in ppm, intensities, etc in the table at the bottom.
The PSM table also features convenient search options to quickly locate the entries of interest.
Sequence coverage
FragmentLab also boasts a feature packed sequence coverage viewer that can be selected through Analyze → Sequence coverage. By loading the FASTA file, FragmentLab will automatically determine the coverage maps for all proteins.
The features showcased in the following order:
1. Three different visualization options;
2. Adaptive spacing for multi-line;
3. Highlight stretch supported by fragments;
4. Amino acid property coloring;
5. Intensity coloring of the peptides;
6. Highlight where the protease cuts.
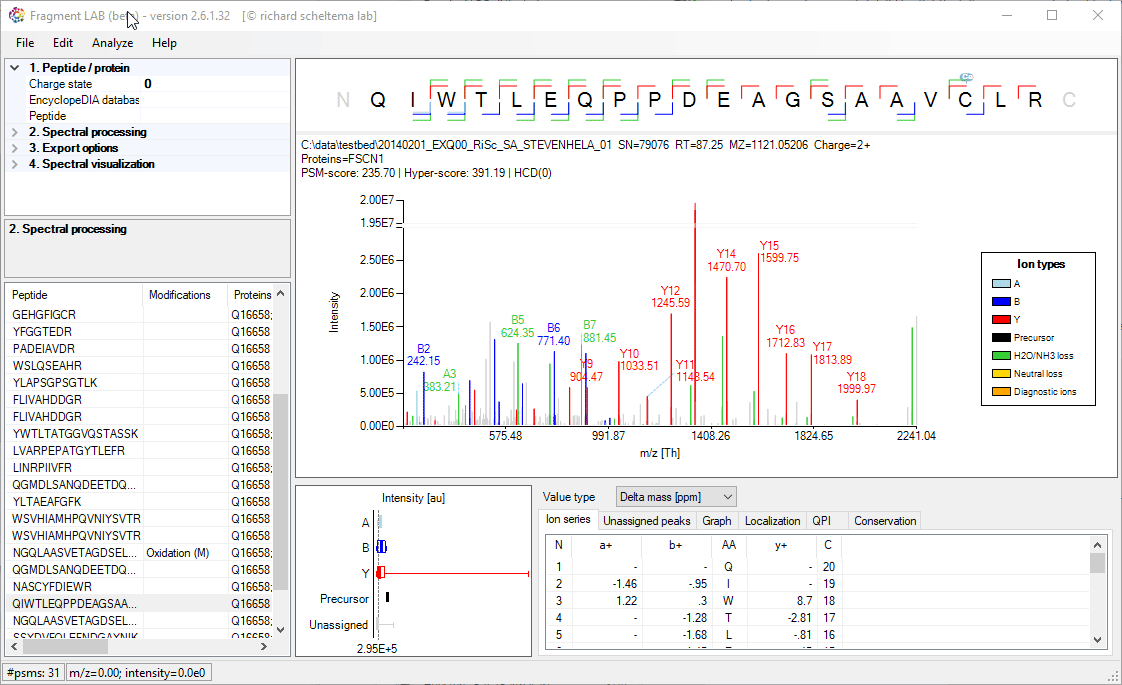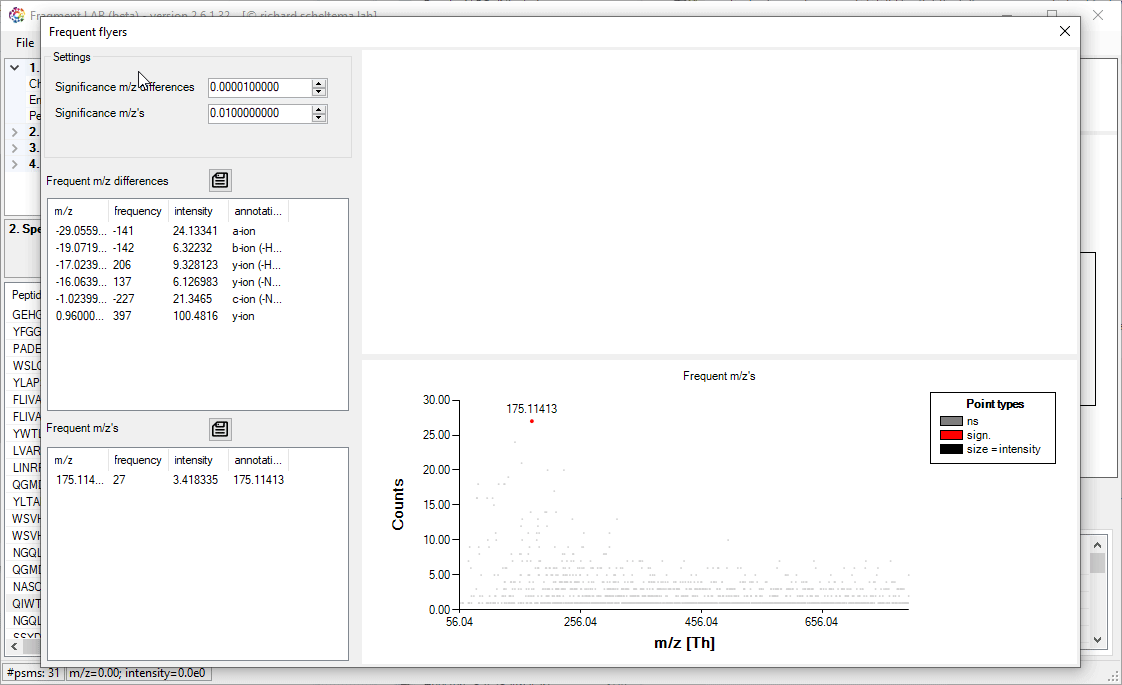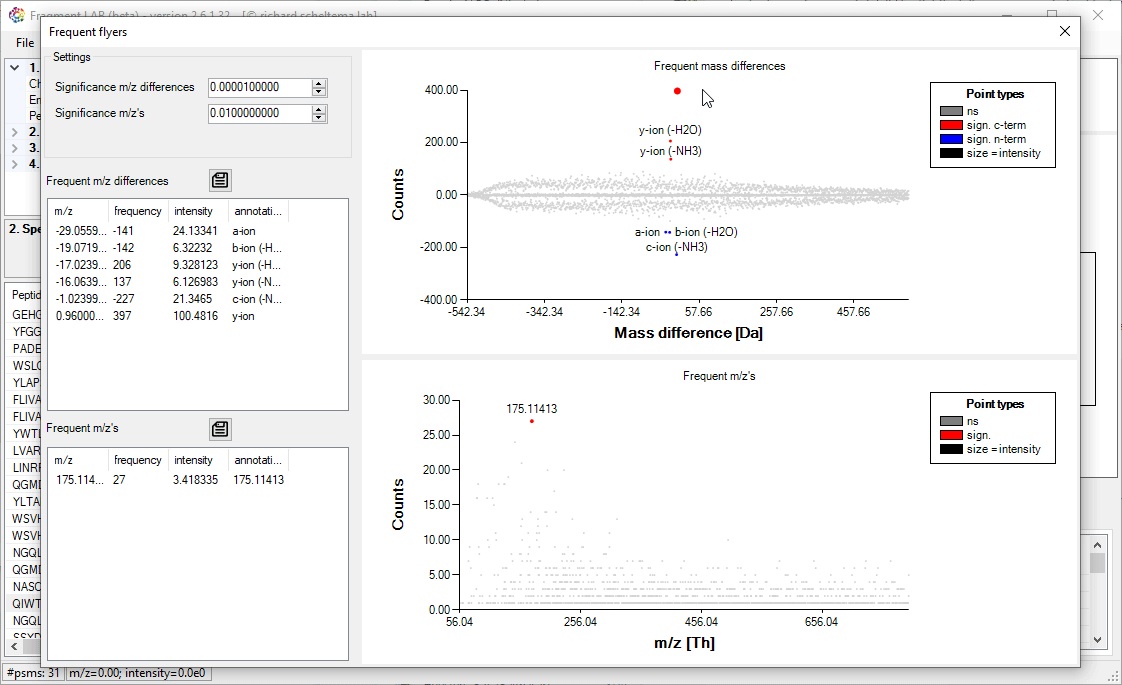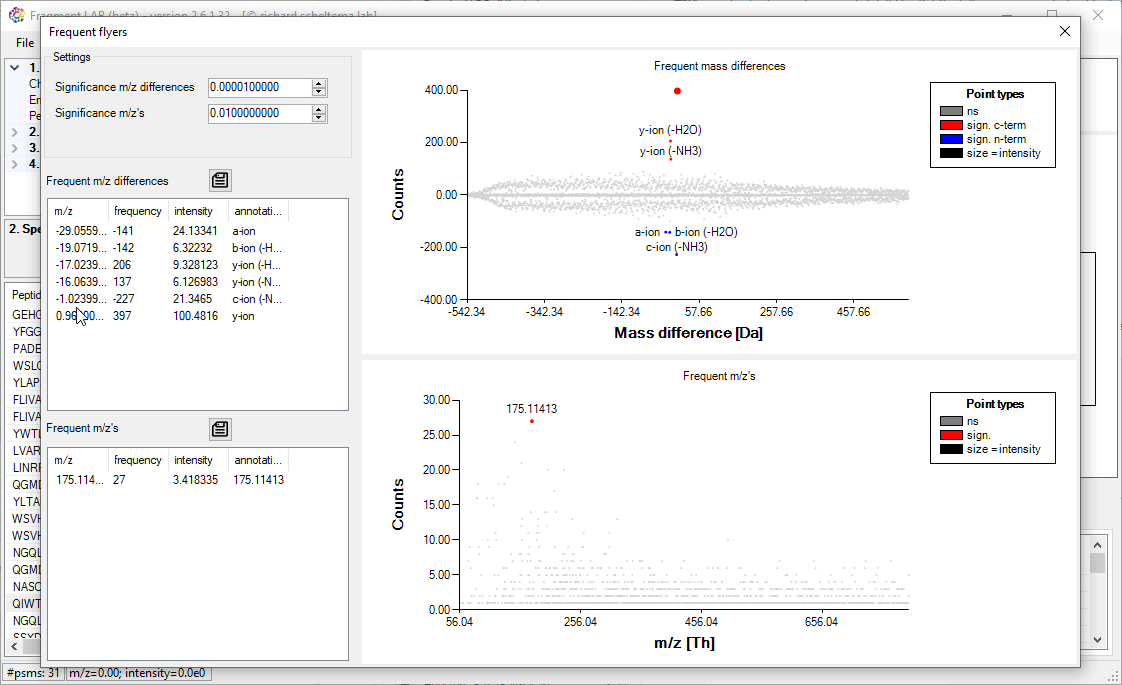
Frequent flyers
An unbiased algorithm to extract, for PTMs, neutral losses and diagnostic ions from a dataset.
For the neutral losses (frequent mass differences) the algorithm investigates all mass differences that frequently occur in relation to the known sequence for each spectrum. This requires knowledge of the peptide identifications out of the search engine, but the results can help to improve the database searches by explaining more of the peaks in the fragmentation spectra.
For the diagnostic ions (frequence m/z’s) the algorithm does not require knowledge of the peptide identity, but simply counts how often a particular m/z exists in the collection of fragmentation spectra.
For both approaches, the known m/z (differences) are annotated. Please note that the exported m/z values are binned and need to be investigated in the spectra to get to the high precision values.
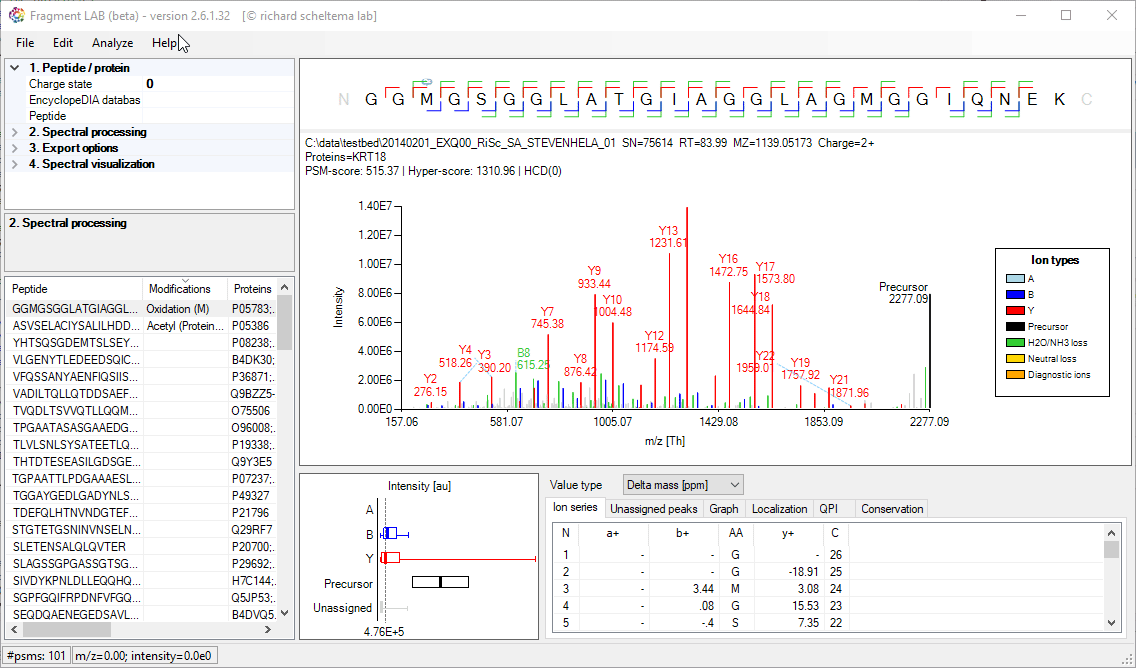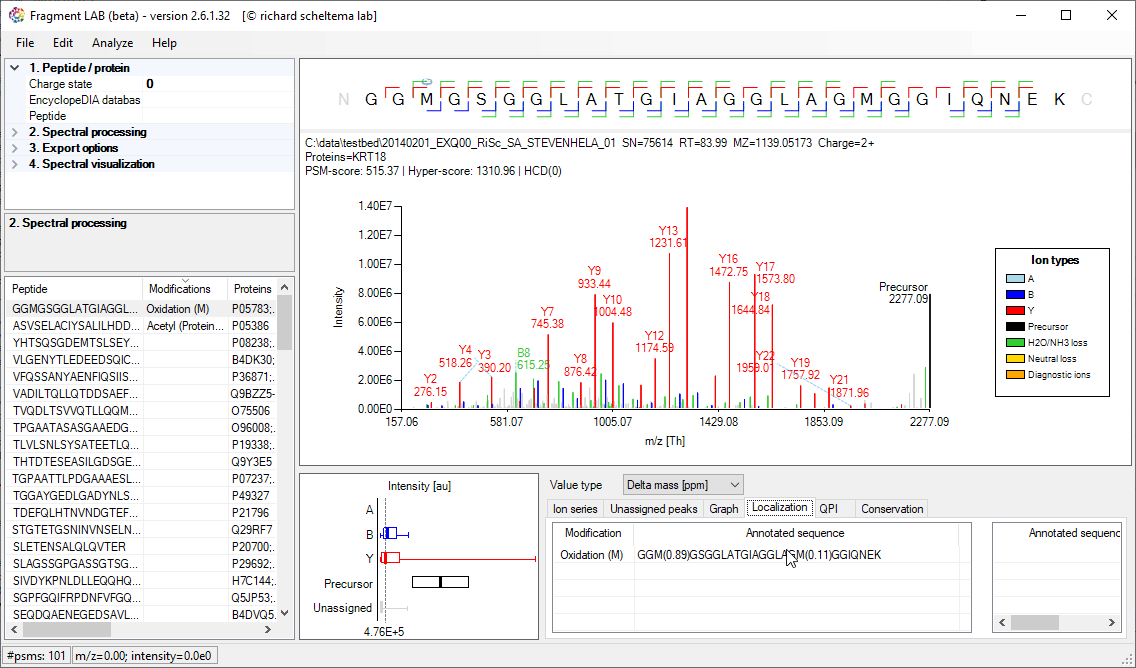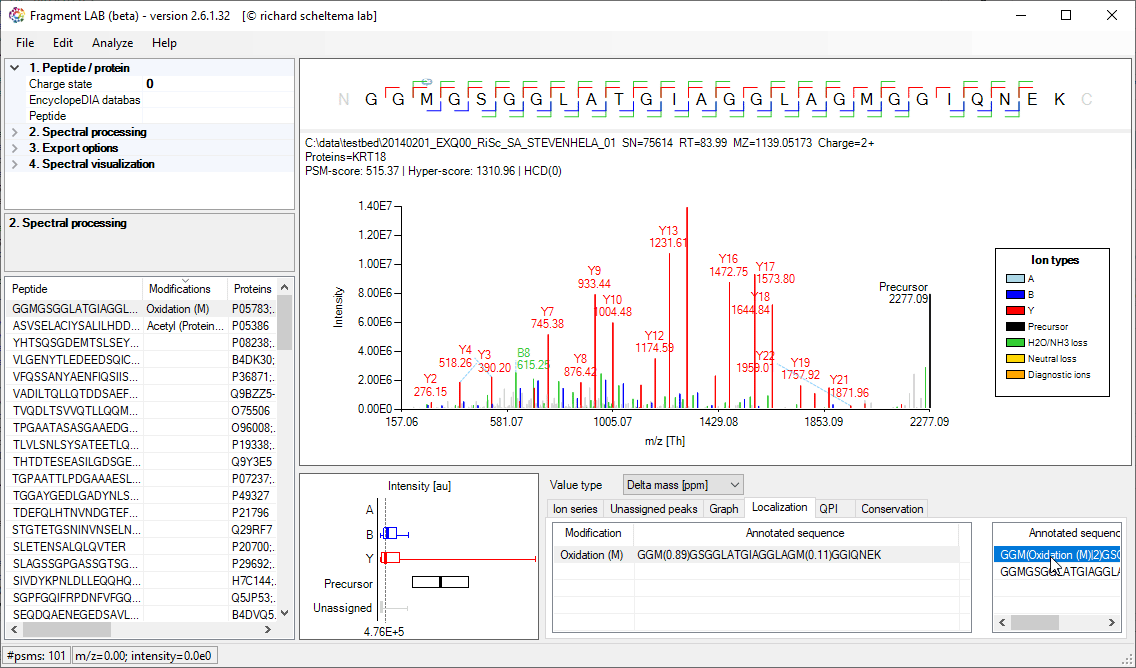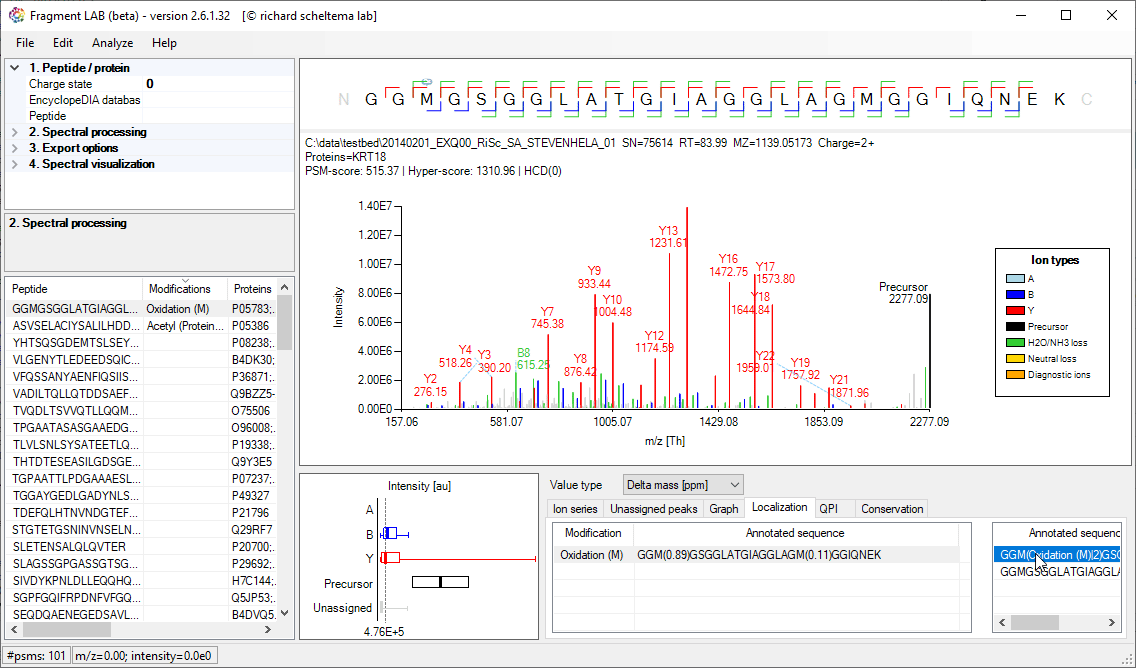
PTM Localization
Localisation of PTMs is generally driven by a localization score, with which search engines try to provide an estimate of how likely the PTM is placed on the indicated residue. Very convenient as it is a single number, but it also hides what the spectrum actually supports.
FragmentLab provides a visual feedback of what happens if the PTM is placed on another location in the peptide sequence. This can be useful for gaining confidence in an identification before proceeding with further experiments on the found site.